Safety and efficacy of remote electrical neuromodulation for the acute treatment of chronic migraine: an open-label study
Brian Grosberg et al, Headache and Orofacial, 2021
Abstract
Introduction: Remote electrical neuromodulation (REN) is an acute treatment of migraine. The results from several studies in patients with episodic migraine suggest that REN is an effective and safe acute treatment of migraine. A recent pilot study provided initial support that REN is effective in patients with chronic migraine as well.
Objectives: The current study aimed to validate and provide further evidence for the safety and efficacy of REN in a large sample of patients impacted by chronic migraine.
Methods: In this open-label, single-arm study, patients with chronic migraine treated their headaches with the REN device (Nerivio, Theranica Bio-Electronics Ltd, Israel) for 4 weeks. Participants used an electronic diary to record their symptoms at treatment initiation, 2 hours after treatment, and 24 hours after treatment. The primary end point was the percentage of subjects who achieved pain relief at 2 hours posttreatment. Secondary end points included pain freedom and improvement of associated symptoms and functional disability.
Results: One hundred twenty-six subjects were enrolled into the study, of which 91 subjects had an evaluable treatment with REN. Pain relief and pain disappearance at 2 hours were achieved by 59.3% (54/91) and 20.9% (19/91) of modified intent-to-treat subjects, respectively (with worst-case sensitivity analysis indicating 54.5% and 19.2%, respectively). Sustained pain relief at 24 hours was observed in 64.4% (29/45) of those who achieved pain relief at 2 hours (with worst-case sensitivity analysis indicating 45.6%). The findings of the study show that REN has a favorable effect on nausea, photophobia, and phonophobia and improves functional ability. One device-related adverse event was reported.
Conclusions: Remote electrical neuromodulation treatments results in the relief of migraine headaches and associated symptoms, thus offering a drug-free acute treatment option for people with chronic migraine. Trial registration: ClinicalTrials.gov NCT04194008.
Keywords: Conditioned pain modulation, Medication overuse headache, REN, Nerivio, Nonpharmacological
1. Introduction
Chronic migraine is characterized by frequent headaches experienced on at least 15 days per month for at least 3 months, of which 8 or more days meet the diagnostic criteria for migraine or respond to migraine-specific acute treatments, such as triptans. Annually, approximately 3% of people with episodic migraine progress to chronic migraine. Risk factors for migraine chronification consist of nonmodifiable risk factors which include older age, female sex, Caucasian race, and genetic factors and modifiable factors which mainly include medication overuse, type of medication used, and high attack frequency.
Treatments of chronic migraine are mainly aimed at reducing the rate of monthly migraine headaches and decreasing the disability associated with migraine without overusing acute medications to avoid the development of medication overuse headache (MOH). The challenge is, thus, to relieve the migraine symptoms of an ongoing attack on the one hand and to reduce acute medication use on the other hand. Nonpharmacological interventions may over- come this unmet need by providing a drug-free treatment option.
New nonpharmacological acute treatments of migraine have emerged during the past decade. Noninvasive neuromodulation technologies for the acute treatment of migraine include remote electrical neuromodulation (REN; Nerivio; Theranica Bio- Electronics Ltd, Netanya, Israel), supraorbital transcutaneous electrical nerve stimulation (Cefaly), noninvasive vagal nerve stimulation (nVNS; gammaCore), and spring transcranial mag- netic stimulation (eNeura).
Remote electrical neuromodulation is a recently developed nonpharmacological acute migraine treatment which noninva- sively stimulates upper arm peripheral nerves. The REN device (Nerivio; Theranica Bio-Electronics Ltd) triggers an endogenic analgesic mechanism named conditioned pain modulation, in which pain in 1 body part is inhibited by pain in another body region.Remote electrical neuromodulation has been previously validated for acute migraine attacks. Several studies confirmed the effectiveness and safety of REN for acute treatment of episodic migraine. A recent pilot study also demon- strated a consistent response to REN in 38 people with chronic migraine. In addition, a recent meta-analysis found REN to be the only migraine neuromodulation intervention for which there is sufficient published high-quality research, and thus the only one for which efficacy was well established.
The current study is an open-label, single-arm safety and efficacy study, aimed to extend these findings in a large sample of patients affected by chronic migraine, to further elucidate the safety and efficacy of REN as an abortive treatment in this population.
2. Methods
2.1. Subjects
Patients were men or women aged 18 to 75 years who were diagnosed with chronic migraine (ie, had between 15 and 23 headache days per month, of which at least8 days per month had a migraine phenotype) according to the International Classifica- tion of Headache Disorders-3 criteria.7 Patients either did not use preventive medications or were on a stable dose of a migraine preventive medication during the 2 months before enrollment and continued using the same medication during the study period. The exclusion criteria were as follows: (1) implanted electrical or neurostimulator device; (2) congestive heart failure, severe cardiac, or cerebrovascular disease; (3) uncontrolled epilepsy; (4) lack of efficacy, after an acceptable experience, of at least 2 migraine-specific acute treatments; (5) pregnancy, nursing, or trying to conceive; (6) other pain, medical, or psychiatric conditions that the investigator deemed as a confounding factor;(7) cannot use smartphones; (8) previous experience with the device; and (9) enrolled into another interventional study.
This study was conducted at 9 U.S. centers (Clinicaltrials.gov NCT04194008). The study was approved by the Western Institutional Review Board (approval No. 20192678) and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all the participants before the start of the study.
2.2. Design and procedures
This trial was a prospective, open-label, single-arm, multicenter study. The study started with a 4-week run-in phase, which was used to assess eligibility based on the number of reported attacks and compliance to report pain levels at baseline and at 2 hours posttreatment. Eligible patients continued into a 4-week treatment phase, in which they were requested to treat all migraine attacks within 1 hour of symptom onset with an individualized stimulation intensity that is just below the pain threshold. Participants were asked to refrain from the use of medications within 2 hours after treatment. Participants used an electronic diary application to record their symptoms at treatment initiation, 2 hours after the treatment, and 24 hours after treatment. The collected data included pain level ratings using a 4-point scale of none, mild, moderate, and severe;presence or absence of nausea or vomiting, photophobia, and phonophobia; and functional disability which was rated on a 4-point scale of no limitation, some limitation, moderate limitation, and severe limitation. Medication use was also recorded at 2 hours and 24 hours.
2.3. Remote electrical neuromodulation device and treatment
The REN device has been described in detail elsewhere.20 In brief, the device is applied to the upper arm for 45 minutes and stimulates C and Aδ noxious fibers using a proprietary electrical signal. For each treatment, the participants were instructed to set the intensity level individually (using simple ± graphical interface on the phone application) so that the stimulation felt strong yet comfortable and not painful.
Participants received standardized detailed hands-on guid- ance on how to use the device and the application. This guidance was given at the study sites by a trained study coordinator (for those who could not arrive at the site because of COVID-19, the same detailed guidance was given over zoom). In addition, all participants received recorded video instructions, available for them at all times, as well as a detailed written manual.
2.4. Outcomes
The primary outcome was the proportion of subjects who achieved pain relief at 2 hours posttreatment, defined as a decrease from severe or moderate pain to mild or no pain or decrease from mild pain to no pain. Additional efficacy outcomes included proportion of subjects who achieved pain freedom (decrease from mild, moderate, or severe pain to no pain) at 2 hours; disappearance of each symptom of nausea or vomiting, photophobia, and phonophobia at 2 hours; sustained pain relief at 24 hours (defined as pain relief that was maintained or improved at 24 hours as compared to pain relief at 2 hours, with no intake of rescue medications in those 24 hours; only subjects achieving relief at 2 hours were included in the analyses); and improvement in functional ability at 2 hours (ie, a 1-grade decrease or more). Consistency of pain response across multiple attacks, defined as the proportion of subjects experiencing 2-hour pain relief in at least half of their treatments, was also assessed (the analysis comprised a minimum of 2 treatments and included all evaluable treatments for each subject). Assessments of associated symptoms were conducted on subjects who experienced the symptom at baseline and reported data at 2 hours. Functional disability was assessed using a 4-point scale (“no limitation,” “some limitation,” “moderate limitation,” and “severe limitation”) rating for the ability to perform usual activities. Subjects with a baseline values of “no limitation” and data at 2 hours were included in the functional disability analyses. Improvement in function was defined as a decrease between baseline and 2 hours of 1 grade or more.
The intention-to-treat (ITT) data set included all subjects receiving the REN device. Safety assessments were conducted on the ITT population. The modified intention-to-treat (mITT) data set included all subjects who completed the treatment period (ie, excluding participants who were excluded or dropped during the intervention period). Sensitivity analysis was conducted on the entire ITT population, assuming in 1 case (worst case [wc]) that all participants who did not complete the treatment period were nonresponders, and in the other case (best case) that all participants who did not complete the treatment period were responders. The International Headache Society guidelines for acute treatment of migraine trials recommend that at least 48 headache-free hours should precede an attack that is included in an efficacy analysis. However, this constraint cannot be applied in patients with chronic migraine who experience more than 15 monthly headache days. Thus, in the current study, the efficacy analyses were conducted on moderate or severe headache that followed a 24-hour period in which mild pain intensity was not exceeded or a mild headache that followed a 24-hour period of pain freedom (ie, qualifying migraine headache).
2.5. Data analysis
The sample size was calculated on the efficacy end point of pain relief at 2 hours posttreatment. Calculations indicated that a sample size of 110 participants would provide 80% power to determine that 60% (±6%) of the participants will achieve pain relief at 2 hours posttreatment. To account for a potential ~10% drop-out rate or missing data, it was determined that the sample size may be increased up to 150 participants. Because of the outbreak of the COVID-19 pandemic, the enrollment stopped with 126 participants, of which 91 provided data and were included in the final analysis set.
Patient demographics and clinical characteristics were sum- marized with descriptive statistics.
The first treatment of each subject was excluded from the efficacy analyses to allow participants to get familiar with the device (training treatment). Efficacy assessments were con- ducted on the first treated qualifying migraine headache with pain data at baseline and at 2 hours after the training treatment (hereby termed test treatment), ie, the test treatment was typically the second treatment. For the consistency of response over multiple migraine headaches, all treated attacks with pain data at baseline and at 2 hours were included (excluding the training treatment). Treatments in which medication was taken before the 2-hour evaluation were classified as failures. In addition, treatments with missing data were not included in the efficacy analyses.
Categorical variables are presented with the number and percentage of patients in each category and 95% confidence intervals (CIs). Continuous variables are presented with mean and SD. Data were analyzed with IBM SPSS statistics software version 25.0. (SPSS Inc, Chicago, IL). All authors had access to the study data.
3. Results
3.1. Patient characteristics
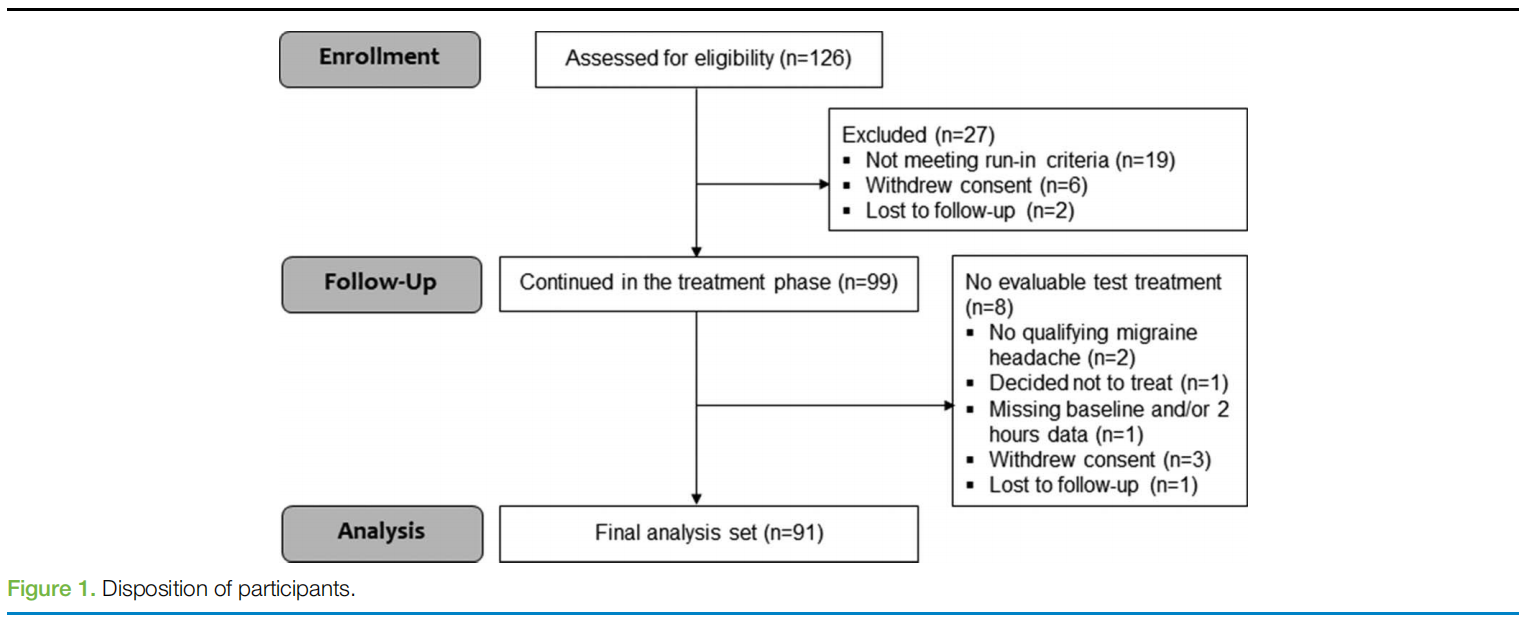
This study was conducted from November 21, 2019, to May 20, 2020 (end of treatment phase). One hundred twenty-six patients were enrolled, of which 6 subjects withdrew consent during the run-in phase (1 patient did not want to travel to the site, 1 patient was unable to comply with protocol requirements, 1 patient did not want to come to the clinic because of the COVID-19 pandemic and a remote visit was not available at that site, and 3 patients did not specify the reason), 2 subjects were lost to follow-up during the run-in phase, and 19 completed the run-in phase but were not eligible to continue according to protocol specifications which require subjects to report at least 6 attacks with data at treatment initiation and at 2 hours after the treatment during the run-in phase. Accordingly, 99 subjects entered the treatment phase and received a device. Five subjects withdrew from the treatment phase (1 patient did not want to continue with the stimulation, 1 patient specified the electrodes were too adhesive, 1 patient experienced connectivity issues because the smartphone was incompatible with the application, 1 patient wanted to start preventive treatment, and 1 patient did not specify the reason). Two subjects were lost to follow-up during the run-in phase. Of those who withdrew consent or were lost to follow-up during the treatment phase, 3 subjects produced sufficient data and were thus included in the final analysis set comprising 91 subjects (Fig.1). In terms of diary compliance, in the run-in phase, 81.6% (829/1016) of sessions had a pain report at both baseline and after 2 hours and in the REN phase, 82.3% (571/694) of sessions had a pain report at both baseline and after 2 hours.
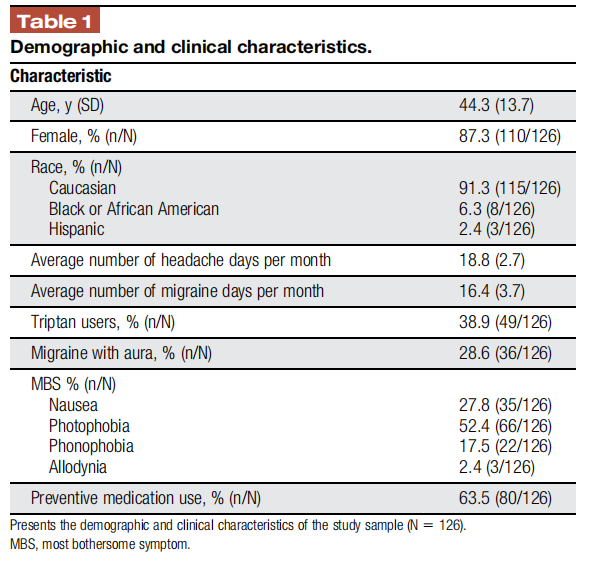
The demographics and clinical characteristics of the 126 subjects enrolled into the study (Table 1) are consistent with published reports on patients with chronic migraine: mainly middle age (mean 44.3 ± 13.7 years, median = 45, interquartile range = 21; lower quartile value = 33, upper quartile value = 54), Caucasian (91.3%) women (87.3%).2 The clinical characteristics of the subjects also correspond with a classification of chronic migraine based on International Classification of Headache Disorders-3 criteria.7 No statistical differences were found in demographics and clinical characteristics between the 126 subjects enrolled into the study and the 91 study completers (see supplementary material, available at http://links.lww.com/PR9/A133 ).
3.2. Treated migraine headaches
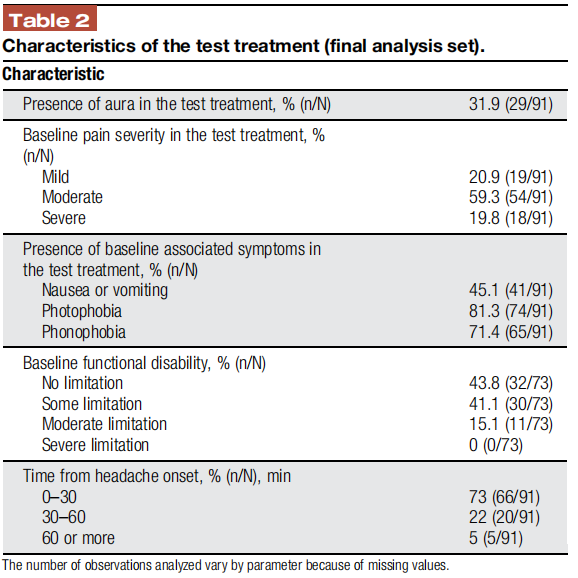
During the treatment phase, 711 REN treatments of qualifying migraine headaches were conducted by 97 subjects. Two subjects did not treat any qualifying migraine attacks (and 3 treated a single attack). The average number of treatments per subject was 7.3 (median 7, interquartile range 5 [lower quartile value = 5, upper quartile value = 10]).
Pain level at treatment start was reported in 635 treatments, of which 81.9% (520/635) were with moderate-to-severe pain intensity. The characteristics of the treated attacks (eg, pain levels and the rate of associated symptoms) were similar to those presented in former migraine studies5,21,22 and accord with attack features of the intended population.8 Table 2 presents the characteristics of the test treatments.
3.3. Efficacy outcomes
3.3.1. Modified intent to treat
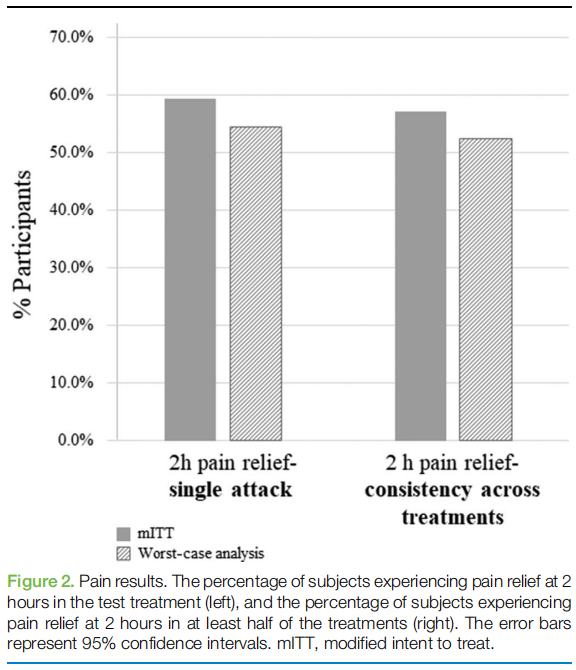
Pain relief and pain-free responses at 2 hours were achieved by 59.3% (54/91; 95% CI 48.5–69.5) and 20.9% (19/91; 95% CI 13.0–30.6) of subjects, respectively (Table 3 and Fig. 2). Pain relief was sustained for 24 hours in 64.4% (29/45) of the subjects (only subjects achieving relief at 2 hours were included in the analyses). Nausea or vomiting, photophobia, and phonophobia disappeared at 2 hours in 48.8% (20/41; 95% CI 32.8–64.8), 40.5% (30/74; 95% CI 29.2–52.5), and 44.6% (29/65; 95% CI 32.2–57.4) of participants, respectively. Furthermore, 59.4% (19/ 32; 95% CI 40.6–76.3) of subjects experienced improvement in functional ability at 2 hours (only subjects with functional disability at baseline were included in the analysis). Consistency analysis demonstrated that 57.1% (52/91; 95% CI 46.3–67.4) of the subjects achieved pain relief at 2 hours in at least half of their treatments (Table 3 and Fig. 2).
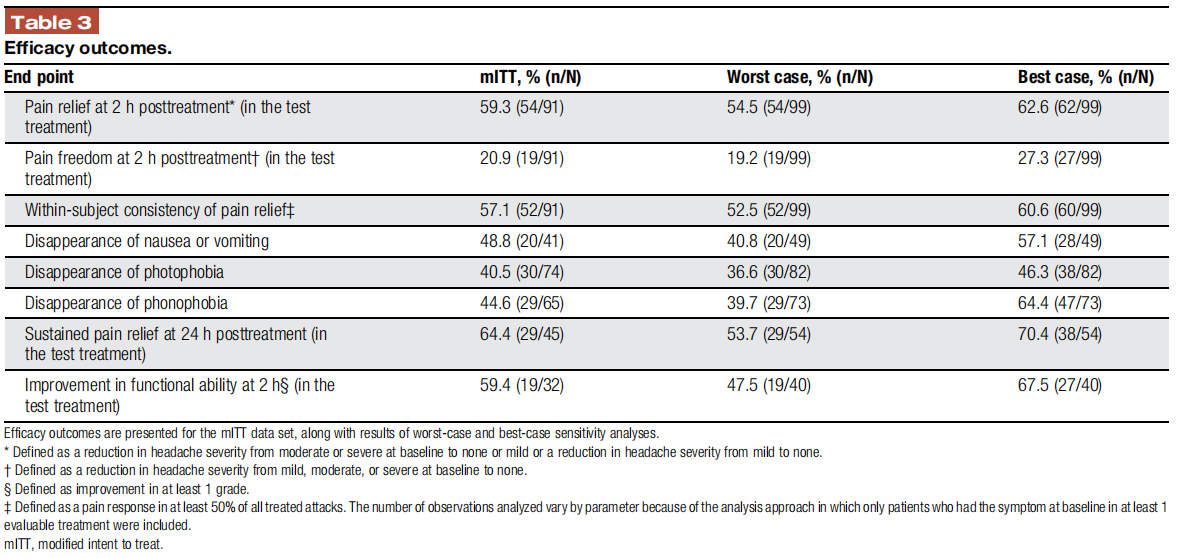
The number of observations analyzed vary by parameter because of the analysis approach in which only patients who had the symptom at baseline in at least 1 evaluable treatment were included.
3.3.2. Sensitivity analysis
Full details on wc and best-case rates of response are presented in Table 3.
3.4. Safety
Ninety-nine subjects who received the device were included in the safety assessments. 9.1% (9/99; 95% CI 4.2–16.6) of subjects experienced at least 1 adverse event, and 1.0% (1/99) of the subjects experienced a device-related adverse event in which pain in the arm was felt after the use of the device on that arm. This adverse event was mild, resolved within 24 hours without medication. The other adverse events which were deemed unrelated to the device included sinus infection (1 patient), upper respiratory infection (2 patients), ear infection (1 patient), viral infection (1 patient), tooth infection (1 patient), leg pain (1 patient), and poison ivy rash (1 patient). No serious device- related adverse events were reported, and no subject withdrew consent because of an adverse event.
4.Discussion
The current study shows that REN provides an effective and safe acute treatment option for people with chronic migraine. The results from studies with episodic migraine showed that REN provides an effective treatment alternative which may reduce acute medication use rates. A recent pilot study has also provided initial support for the effectiveness of REN treatmentsin chronic migraine, although the sample size in that study was small. To further explore the efficacy of REN in the chronic migraine population, the current study was conducted in a large population of patients with chronic migraine. Treatments with REN in this population resulted in a clinically meaningful pain response, relief in the associated symptoms, and improved ability to function.
To provide a full view of the results, we list herein the results of the mITT (ie, results of all participants who completed the intervention phase), followed by a wc sensitivity analysis in parentheses (ie, an analysis assuming that all participants who dropped out during the intervention phase were nonresponders to all parameters regardless of the reason for drop out).
Nearly 60% (wc 54.5%) of the subjects experienced relief of pain at 2 hours, and approximately 65% (wc 53.7%) of subjects had sustained pain relief at24 hours. The pain response observed in this study is also similar to findings reported in an open-label study which evaluated the efficacy of a vagus nerve stimulation device (gammaCore) in 34 people with chronic migraine, in which the proportion of subjects reporting 2-hour pain relief was 58.8%. Remote electrical neuromodulation treatments also had a favorable effect on nausea or vomiting, photophobia, and phonophobia. Furthermore, approximately 60% of patients achieved improvement in function at 2 hours.
The frequent nature of chronic migraine requires repeated treatments, and thus acute treatment should be consistently effective and tolerable over multiple migraine attacks. Consis- tent with previous results, our findings show that 57.1% (wc 52.5%) of the patients achieved pain relief at 2 hours in at least half of their attacks, indicating that REN may overcome an unmet need for a consistent acute therapy.11 Although no head-to-head trials were performed and variance in the method, design, and studied population (episodic vs chronic) is apparent, compari- sons of these results indicate that the consistent response to REN is similar to that of triptans (57% for REN vs 47%–72% for triptans) and may be superior to nVNS (57% for REN vs 47% for nVNS). Given that 8 of 99 participants did not complete the intervention phase, results should be viewed as a range, rather than a single number. We thus discuss mITT results along with a wc sensitivity analysis, and both should be considered when evaluating the results. It should be noted however that reasons for noncompletion varied and included individuals who were excluded from the analysis because of lack of qualifying migraine attacks (as per the protocol; eg, did not have 15 headache days per month), as well as individuals who withdrew consent, decided not to treat, or did not report pain levels.
The current investigation also demonstrates that REN main- tains favorable safety and tolerability profiles. The adverse events found in this study are comparable with the known tolerability profile of REN. There was a low rate of device-related adverse events (1.0%), and there were no serious device-related adverse events reported. For MOH, it has been recently shown that incorporating Nerivio into usual care may reduce medication use and thus may reduce the risk for MOH.Furthermore, neuro- modulation is considered a preferable treatment for individuals with chronic migraine to prevent escalation to MOH. However, the direct effects of REN on MOH were not tested in the current study (or any other study to date), and thus any effect, positive or negative, cannot be evaluated based on the current data set.
It should be noted that the results of the REN treatment were not compared with those of sham stimulation, which may be considerable. Yet, comparing the mITT results to an estimated 2- hour pain relief sham response of 38.8% observed in previous studies of REN shows a statistically significant and clinically meaningful therapeutic gain (20.5%; P=0.004), and thus placebo and nocebo effects, while no doubt exist, are not likely to explain the current results. An additional limitation is the small number of subjects included in some of the evaluated parameters which stems from the analysis approaches in which only patients with a specific symptom reported as present at baseline are included and due to missing data. Therefore, additional studies in a larger number of subjects are needed. Finally, data from 91 participants were included in the analysis data set, whereas 126 participants were recruited to the study. Data from 35 participants were not included (as detailed in the disposition figure) primarily because of nonadherence with the study’s protocol. Importantly, 27 of the 35 participants were excluded in the run-in phase, in which the REN device was not yet given to the participants and was not used or tested. This phase was designed particularly for this purpose, ie, to assess eligibility based on the number of reported attacks and compliance to report pain levels at baseline and at 2 hours posttreatment, regardless ofthe REN intervention. Ninety-nine participants entered the treatment phase (and received a REN device), of which 8 did not complete their participation in this phase (as detailed in the disposition chart). Those 8 are within the anticipated 10% of drop out or data loss. Regarding the rate of pain freedom(20.9%), although the rate of pain freedom is slightly lower than that of some medications, REN offers a superior safety profile, with nearly no side effects (1.0% adverse events in the current trial), ie, in comparison to the risk of MOH, as well as gastrointestinal symptoms and other severe side effects associated with standard care medications for migraine. In addition, as discussed, the pain relief rates are comparable with those of triptans and may be superior to nVNS.
In addition, although this has no bearing on the results or interpretation, it should be mentioned that the study was registered in clinicaltrials.gov in December, whereas enrollment started in November 2019 (the first participant received their REN device only after registration, no intervention was performed before registration nor was any decision taken or could have been taken during this strictly observational period).
In conclusion, this study supports the efficacy and positive benefit-to-risk ratio for REN used for the acute treatment of chronic migraine. This investigation demonstrates that REN may provide an alternative nonpharmacological acute treatment option in people with chronic migraine, holding the potential to diminish medication use in a population prone to develop MOH.
