Remote electrical neuromodulation for acute treatment of migraine in adolescents
Andrew D. Hershey et al, Headache, 2020
ABSTRACT
Objectives: Migraine is a common disabling neurological disorder. Current acute treatments for migraine in adolescents are mostly pharmacological and may have limited effectiveness, can cause side effects, and may lead to medication overuse. There is an unmet need for effective and well-tolerated treatments. Remote electrical neuromodulation (REN) is a novel acute treatment of migraine that stimulates upper arm peripheral nerves to induce conditioned pain modulation (CPM)—an endogenous analgesic mechanism. The REN device (Nerivio®, Theranica Bio-Electronics Ltd., Israel) is a FDA-authorized device for acute treatment of migraine in adults. This study assessed the efficacy and safety of REN in adolescents with migraine.
Design and Methods: This was an open-label, single-arm, multicenter study in adolescents (ages 12–17 years) with migraine. Participants underwent a 4-week run-in phase. Eligible participants continued to an 8-week treatment phase with the device. Pain severity, associated symptoms, and functional disability were recorded at treatment initiation, and 2 and 24 hours post-treatment. The primary endpoints of this study were related to the safety and tolerability of REN. The secondary endpoints were related to device efficacy and included the proportion of participants who achieved pain relief at 2 hours post-treatment and the proportion of participants who achieved pain freedom at 2 hours. The presented results reflect an interim analysis with subsequent stopping of the rest of the study.
Results: Sixty participants were enrolled for the study; of these, 14 failed to meet the run-in criteria and 1 was lost to follow-up. Forty-five participants performed at least one treatment, of which 39 participants completed a test treatment with REN. One device-related adverse event (2%) was reported in which a temporary feeling of pain in the arm was felt. Pain relief and pain-free at 2 hours were achieved by 71% (28/39) and 35% (14/39) participants, respectively. At 2 hours, 69% (23/33) participants experienced improvement in functional ability.
Conclusion: REN may offer a safe and effective non-pharmacological alternative for acute treatment in adolescents.
KEYWORDS
acute migraine treatment, adolescents, headache, medication overuse headache, Nerivio, remote electrical neuromodulation
Introduction
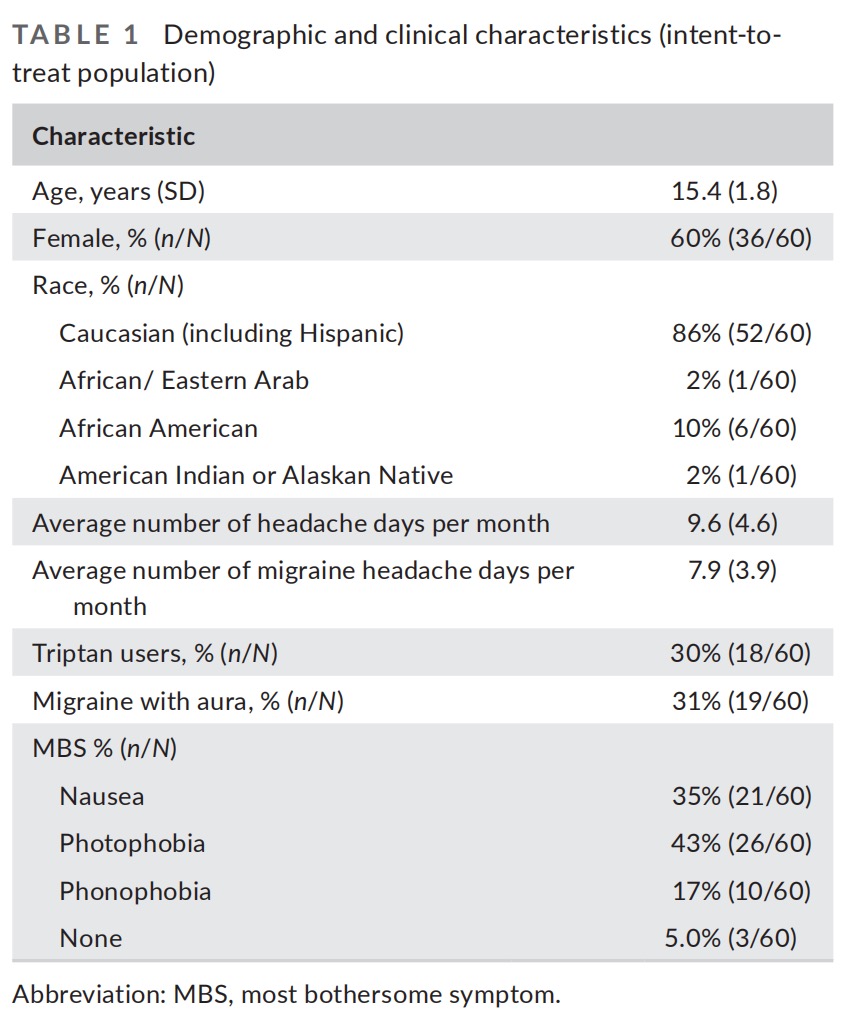
Migraine is one of the most prevalent and disabling diseases worldwide, affecting approximately 9% of children and adolescents. The prevalence of migraine increases with age, particularly during adolescence. Migraine in adolescents has been associated with missed school days, poorer performance in school, negative effect on peer and social interactions, and negative impact on quality of life.
The majority of current migraine acute treatments for adolescents are pharmacological. These treatments may not always be effective, they can cause side effects, and their overuse may lead to medication overuse headache, and migraine chronification. Thus, there is a great unmet need for alternative acute migraine treatments that are both effective and well tolerated to improve the health and quality of life of adolescents with migraines.
Noninvasive neuromodulation devices represent an emerging field in the acute treatment of migraine. Remote electrical neuromodulation (REN) is a non-pharmacological, noninvasive, acute migraine treatment that stimulates upper arm peripheral nerves to induce conditioned pain modulation—an endogenous analgesic mechanism in which a conditioning stimulation inhibits pain in remote body regions. The REN device (Nerivio®, Theranica BioElectronics Ltd., Israel) is a wireless, wearable, battery-operated stimulation unit controlled by a smartphone software application. The device is applied for 45 minutes to the lateral upper arm and mainly stimulates small skin nerves.
The safety and efficacy of REN have been previously assessed for migraine in a randomized, double-blind, sham-controlled multicenter study conducted on adults aged 18 years and above. 12 This study demonstrated that REN provides superior clinically meaningful relief of migraine pain and most bothersome symptom (MBS) compared to placebo (pain relief: 66.7% vs. 38.8%), offering a safe and effective non-pharmacological alternative for acute migraine treatment. The aim of this study was to evaluate the safety and efficacy of REN for acute treatment of migraine in adolescents. We hypothesized that the safety of the REN device in adolescents will be favorable and similar to that observed in adults.
METHODS
Standard protocol approvals, registrations, patient assents, and parents/guardian consents
The study protocol was reviewed and approved by the appropriate institutional review board for each site and was conducted according to Good Clinical Practice and the Declaration of Helsinki guidelines. Before undergoing any study procedures, patients provided written informed assent and their parents/guardian provided written informed consent. The study is registered with ClinicalTrials.
Study design and participants
Adolescents with migraine with or without aura participated in this prospective, open-label, single arm, multicenter study conducted at 12 sites in the USA. Patients were eligible to participate if they were 12–17 years old at the time of informed consent (inclusive), met the International Classification of Headache Disorders, 3rd edition crite- ria for migraine with or without aura, had history of at least three migraine attacks per month for each of the 2 months preceding study enrolment with any number of headache days per month (i.e., the study included both patients with episodic migraine and patients with chronic migraine), reported typical headache duration of at least 3 hours (when untreated or unsuccessfully treated), and were on either no, or stable migraine preventive medications in the last 2 months prior to recruitment. Exclusion criteria were: (a) pregnancy, nursing, trying to conceive, (b) implanted electrical and/or neuro- stimulator device, (c) congestive heart failure, severe cardiac or cer- ebrovascular disease, (d) epilepsy, (e) use of cannabis 1 month prior to enrollment, (f) undergoing nerve block in the head or neck within the last 2 weeks, (g) treatment with onabotulinumtoxinA (Botox) to the head and/or neck for 3 months before enrollment and/or dur- ing the study, (h) any history of anti-calcitonin gene-related peptide antibody treatment, (i) pure menstrual migraine, (j) parenteral treat- ments for migraine within the previous 2 weeks, (k) other significant illness that in the opinion of the investigator may confound the study assessments, (l) unable to use a smartphone, (m) previous experience with the device, (n) participating in any other interventional clinical study, and (o) arm circumference below 7.9 in.
The REN device
The REN device is a wireless, wearable, noninvasive stimulation device applied to the lateral upper arm between the bellies of the lateral deltoid and the triceps for 45 minutes. The device stimulates small skin nerves using a proprietary electrical signal comprising a modulated, symmetrical, biphasic, square pulse with a modulated frequency of 100–120-Hz, pulse width of 400 μs, and up to 40 mA output current (adjusted by the patient). The pulse is designed to stimulate C and Aδ noxious sensory fibers above their depolarization thresholds, yet the stimulation energy is low enough to maintain the overall sensory experience below the perceptual pain threshold.
Since REN induces a global pain inhibition mechanism, the device can be used on either arm independently from the side of a unilateral headache.
Procedures
After enrollment, participants were trained to use the electronic diary application, installed on their smartphones, and then completed a 4-week run-in phase. Participants who did not have at least three migraine attacks or did not report the pain level at 2 hours post-treatment in at least 66.7% of the attacks were excluded from the study. Eligible participants continued to an 8-week treatment phase. During this visit, participants and their parents/guardian were trained to use the REN device, including finding the optimal intensity level (perceptible but not painful). Participants were instructed to use the device at home for the treatment of four qualifying migraine attacks during a period of up to 8 weeks. A qualifying migraine attack is defined as a migraine attack that: (a) was not preceded by another migraine or other headache within the preceding 24 hours, (b) was not preceded by the use of specific or nonspecific acute migraine medications within the previous 24 hours, and (c) occurred in a setting in which the patient could properly administer the treatment within 60 minutes of onset and complete the migraine diary at 2 hours.
Participants were instructed to avoid taking rescue medications within 2 hours post-treatment. Pain scores (none, mild, moderate, or severe), absence/presence of associated symptoms (nausea/vomiting, photophobia and phonophobia), and functional disability were recorded at baseline, and 2 and 24 hours post-treatment. To assess functional disability, participants recorded at baseline, and 2 and 24 hours post-treatment their response to the following question in their diary: “How do you rate your ability to do school-work or perform your usual activities?” using a 4-point scale (“as usual,” “some ability,” “a little ability,” and “no ability at all”).
Participants who did not achieve satisfactory relief at 2 hours or had headache recurrence could treat again with the device or with usual care. Adverse events reported throughout this phase of the study were recorded.
Outcomes
The primary endpoints of this study were related to the safety and tolerability of REN. Safety was assessed by the incidence of adverse events in general and by seriousness, severity, and association to the device. Treatment tolerability was assessed by the percent of subjects who fail to complete the study because of adverse events. The secondary endpoints were related to device efficacy and included the proportion of participants who achieved pain relief at 2 hours post-treatment, defined as improvement from severe or moderate pain to mild or none, or improvement from mild pain to none; proportion of participants who achieved pain-free (improvement from mild, moderate, or severe pain to none) at 2 hours, and disappearance of associated symptoms (nausea/vomiting, photophobia, and phonophobia) at 2 hours. Exploratory endpoints included sustained pain relief at 24 hours, sustained pain-free at 24 hours, and improvement in functional ability at 2 hours (defined a reduction of at least one grade). Within-subject consistency of pain relief and pain-free responses, defined as the proportion of participants achieving pain relief/pain-free at 2 hours in at least 50% of their treated headaches, were also assessed.
Additional outcome was improvement in migraine-related disability, assessed by the Pediatric Migraine Disability Assessment (PedMIDAS) questionnaire, which asks how migraine interfered with school and daily activities. The questionnaire was administered at baseline (enrollment visit) and at the end of the treatment phase. Although typically the PedMIDAS asks about the last 3 months, in this study participants were asked to refer to the last 2 months since the REN device was used for 8 weeks and the aim was to assess its effect on disability within the intervention period.
Data analysis
This is the primary analysis of the data obtained from this study. The sample size was calculated on the efficacy endpoint of pain relief at 2 hours. Initial calculations show that a sample size of 110 par- ticipants would provide 80% power to determine that 60% (±6%) of the participants will achieve pain relief at 2 hours. To account for a potential ~15% drop-out rate and/or missing data, it was determined that the sample size may be increased to up to 130 participants. Although the power calculation was conducted on the secondary efficacy endpoint, it also accounts for the safety and tolerability primary endpoints, for which a smaller sample size was estimated to suffice to ensure that the majority of safety and tolerability is- sues would be revealed, as typical with device studies conducted on adolescents following a pivotal adult study. The power calculation was, thus, conducted on the pain relief outcome to make sure the study will not be underpowered study to assess the efficacy endpoints, which are an important aspect of the objective of the study even though they were defined as secondary endpoints. Due to the outbreak of the coronavirus disease 2019 pandemic in the United States in March 2020, all study sites stopped enrolling patients to the study. An interim analysis was conducted to assess the statistical power of the data accumulated from the 60 patients enrolled which is approximately 50% of the -planned sample size. Based on the results of this analysis, the data monitoring committee determined that the study can be deemed completed for benefit since pain relief at 2 hours was achieved by more than 60% of participants. Stopping enrollment with a final analysis set of ~40 participants is further supported by an additional power analysis in which the margin of error was increased from 6% in the initial calculation to 10%. Calculations show that a sample size of 40 participants provides statistical power of 80% to determine that 60% (±10%) of the participants will achieve pain relief at 2 hours.
The intention-to-treat population included all participants who received the device and was used for the efficacy and safety analyses. The first reported treatment of each participant was con- sidered a training treatment and was only included in the safety analyses. The efficacy evaluation was based on the first treated qualifying migraine headaches with baseline and 2 hours assessment following the training treatment (hereby termed test treatment). The use of rescue medication before the 2-hours assessment was considered a treatment failure. Treatments with missing data were ex- cluded from all analyses.
For the associated symptoms outcomes, patients with presence of a symptom at baseline and data at 2 hours are included in the anal- yses. For functional disability endpoints, all patients with baseline values of "some limitation," "moderate limitation," or "severe limitation," and data at 2 hours were included in the analyses.
For continuous variables, mean and standard deviation are pro- vided. The assumptions of normality of the age and PedMIDAS variables were verified using the normal quantile–quantile (Q–Q) plot. Interquartile range of these variables is also presented. The analyses of the categorical variables focused on estimating the event rates with corresponding uncertainty, and there were no formal statistical tests and no null hypothesis testing. In this analysis, the number and percentage of patients in each category are provided with 95% confidence intervals calculated using the binomial (Clopper–Pearson) exact method. Data were analyzed with IBM SPSS statistics software version 25.0. (SPSS Inc., USA).
RESULTS
Participants
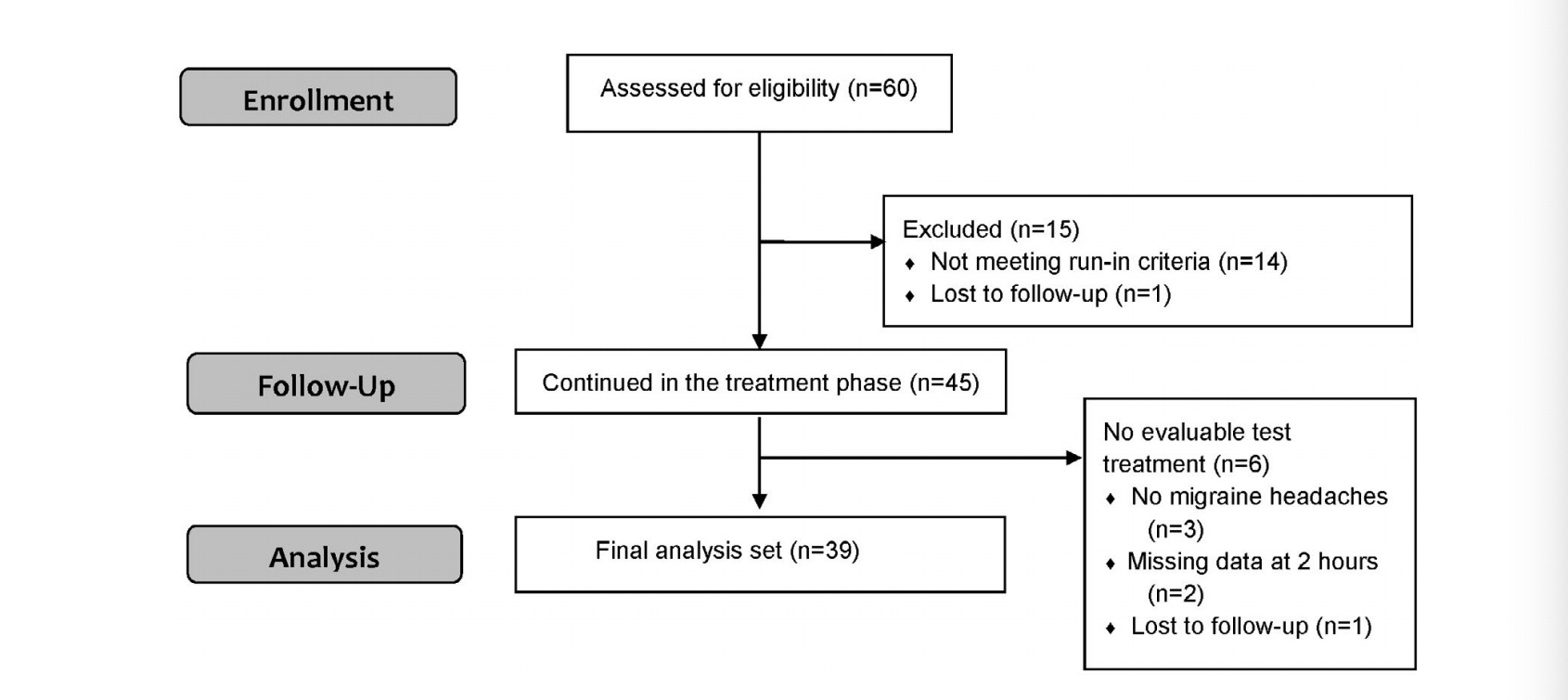
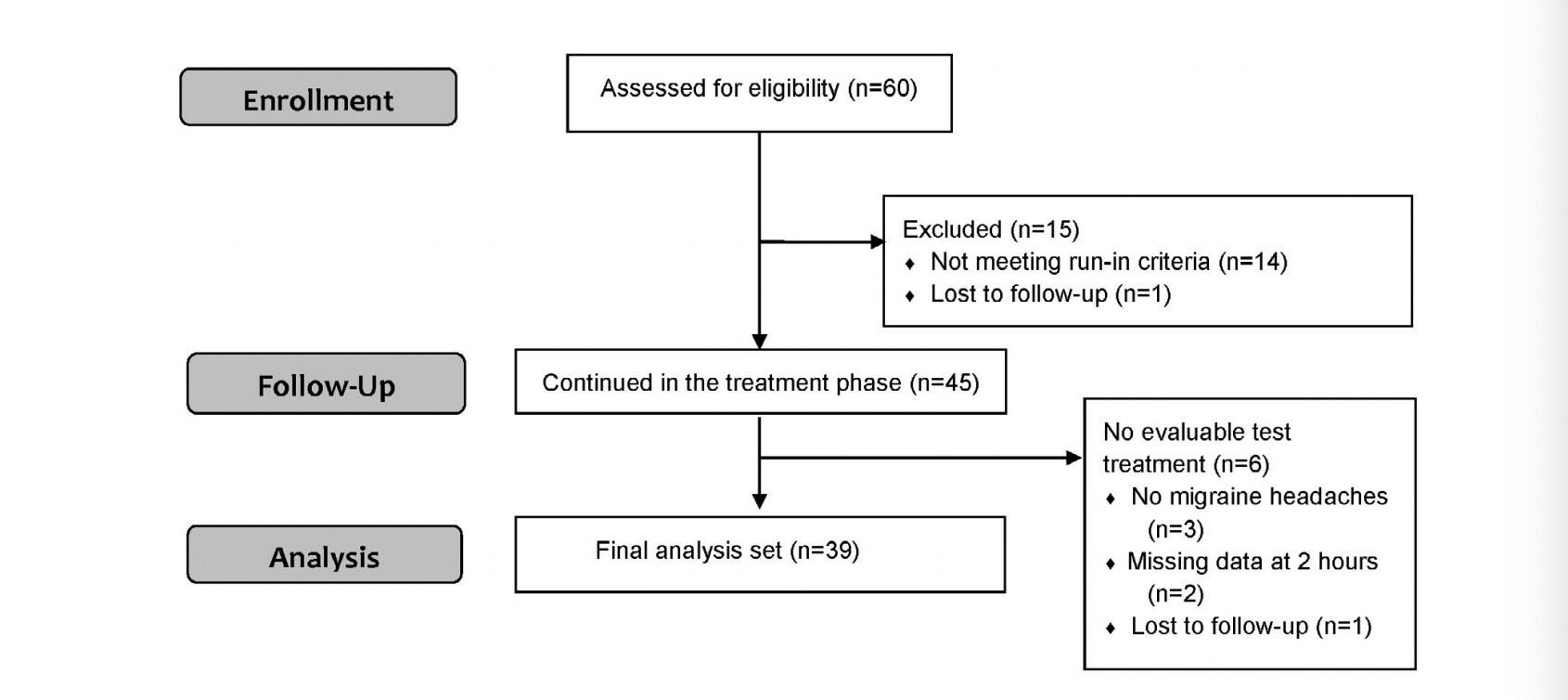
This study was conducted from October 9, 2019 to May 24, 2020 (completion of the treatment phase). A total of 60 participants were enrolled, of which 1 participant was lost to follow-up during the run-in phase, and 14 completed the run-in but were not eligible to continue according to protocol specifications (Figure 1). Of the 14 participants who failed to meet the run-in criteria, 12 participants were ineligible due to insufficient number of attacks (less than three attacks), and 2 participants were ineligible due to noncompliance with migraine attack diary reporting requirements.
Among the 45 participants who entered the treatment phase, all completed at least one treatment (the training treatment) and 39 participants completed at least one additional treatment with base- line and 2-hour data following the training treatment, forming the final analysis set (2 participants had missing data at 2 hours, three participants did not have migraine headaches and one participant was a lost to follow-up; Figure 1).
The demographic and clinical characteristics of the participants (Table 1) and the characteristics of treated migraine headaches were comparable to those reported in previous studies of migraine in adolescents.
Treated migraine headaches
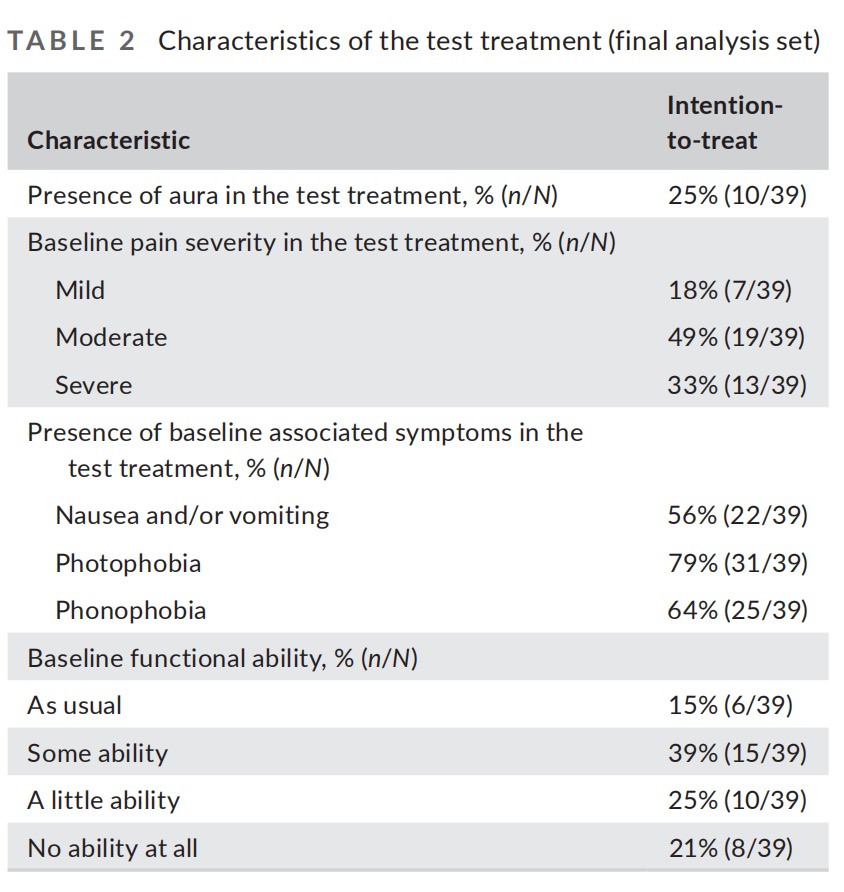
A total of 159 qualifying migraine headaches were treated with the device for which pain data was recorded at baseline and at 2 hours (average of 3.5 treatments per participant). Pain severity of treated migraine headaches was mostly moderate (48% [77/159]). Generally, the characteristics of treated migraine headaches were comparable to those reported in previous migraine studies in adolescents. The characteristics of the test treatments are presented in Table 2.
Safety and tolerability
Safety analyses were performed on all 45 participants who used the device at least once. 10 participants (22%; CI95% 11%–37%) reported at least one adverse event. There was one mild device-related adverse event reported (2%; CI95% 0.06%–11%) in which a temporary feeling of pain in the arm was felt but resolved after the treatment without requiring intervention. The other adverse events which were deemed unrelated to the device included common cold (1), chest congestion (2), influenza (2), leg pain (1), streptococcus pharyngitis (1), upper respiratory infection (1), and worsened migraine (1). There were no device-related serious adverse events and none of the participants withdrew from the study due to device-related adverse events.
Efficacy outcomes
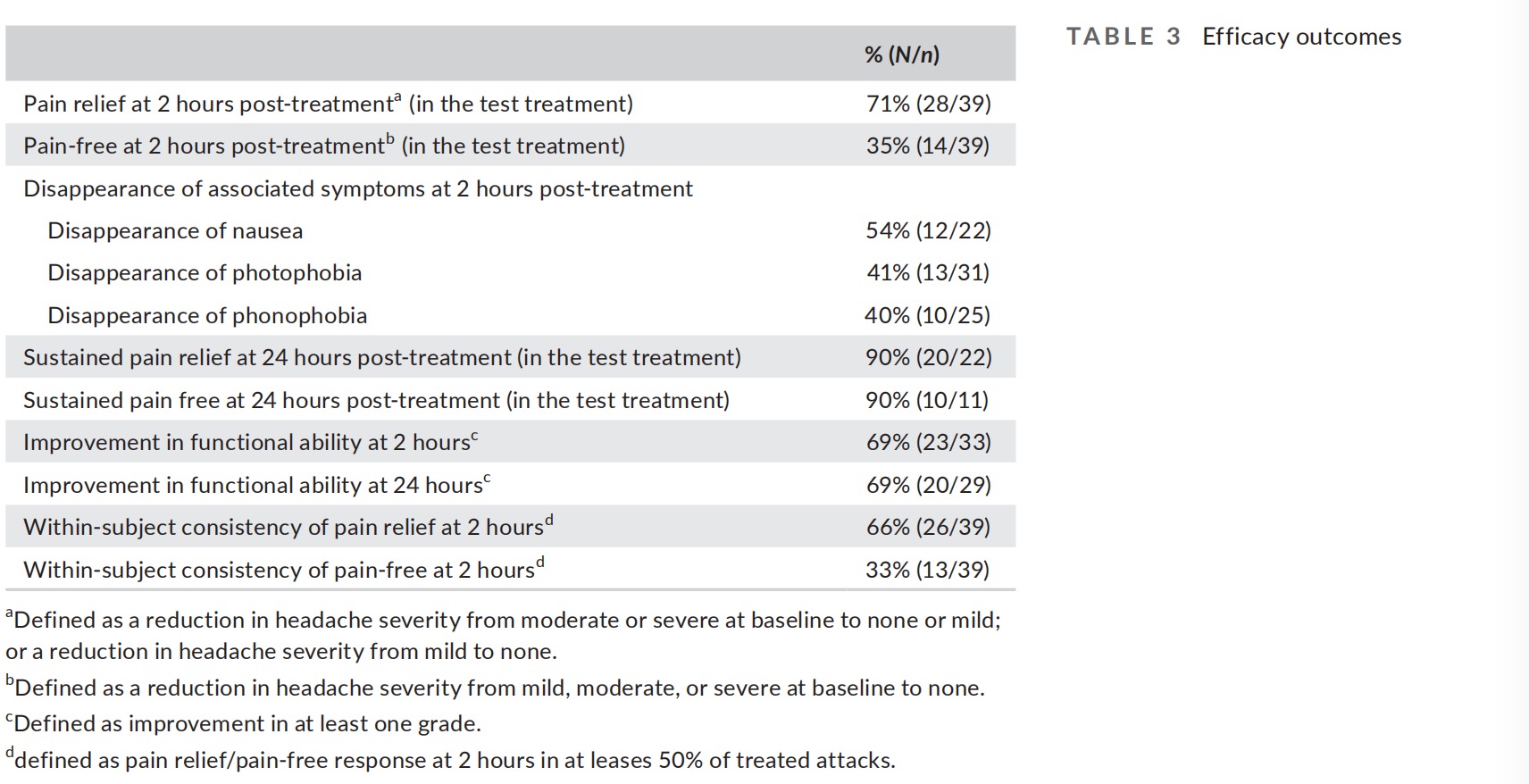
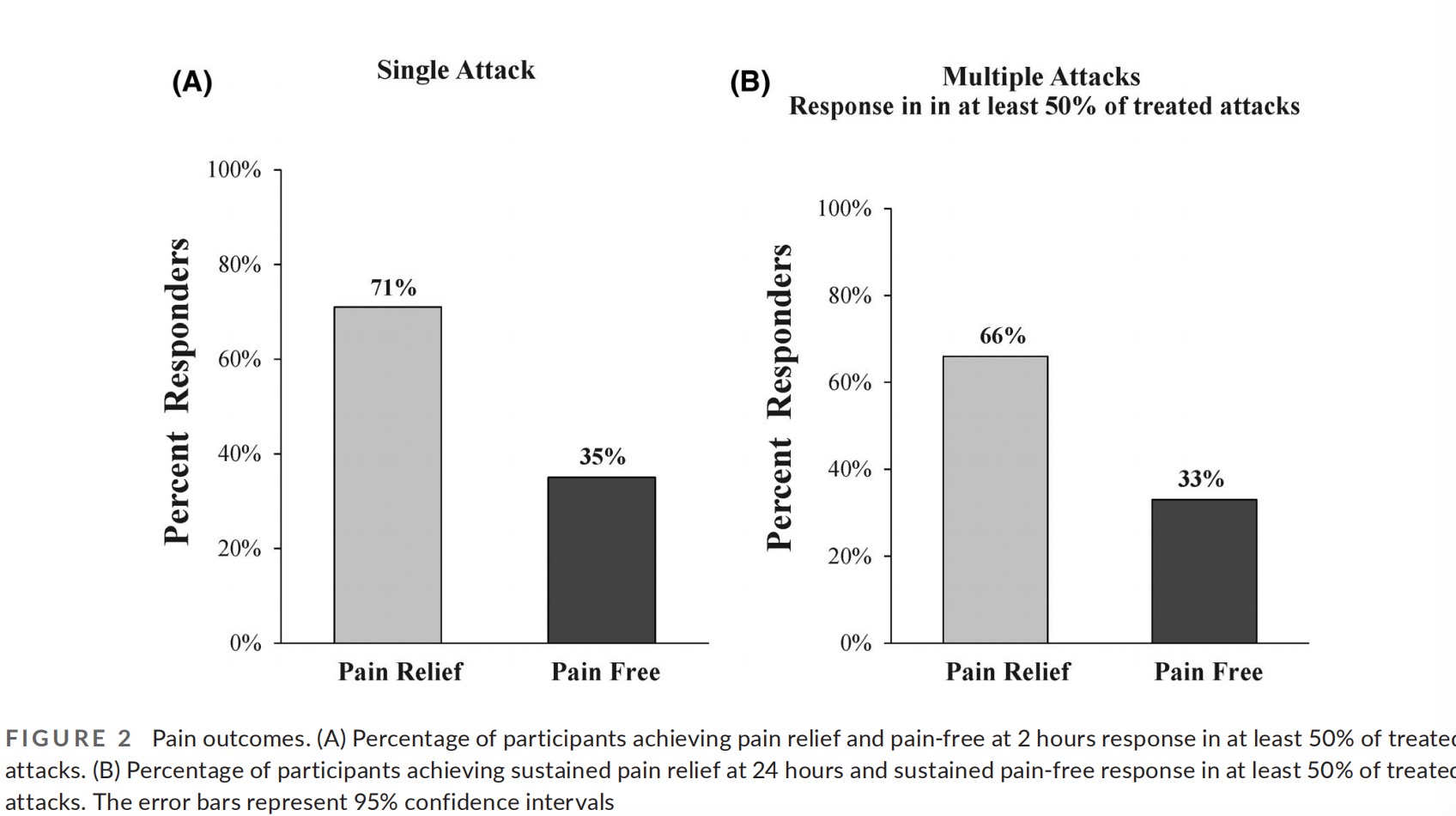
Pain relief and pain-free at 2 hours were achieved by 71% (28/39; CI95% 55%–85%) and 35% (14/39; CI95% 21%–52%) participants, re- spectively (Table 3 and Figure 2). In a sensitivity analysis assuming all treatments with missing pain level data were considered failures, pain relief was achieved by 68% (28/41; CI95% 51%–81%) of the participants.
Pain relief was sustained for 24 hours in 90% (20/22; CI95% 70%–98%) of the participants, and pain freedom was sustained for 24 hours in 90% (10/11; CI95% 58%–99%) of the participants (only subjects achieving relief/freedom at 2 hours were included in the analyses; six participants who achieved pain relief at 2 hours did not report pain level at 24 hours and were excluded from the sustained pain relief analysis and four participants who achieved pain freedom at 2 hours did not report pain level at 24 hours and were excluded from the sustained pain freedom analysis). A sensitivity analysis assuming all treatments with missing pain level data at 24 hours had return of pain (i.e., considered failures) further indicated a favorable sustained response; sustained pain relief was achieved by 71% (20/28; CI95% 51%–87%) of the participants and sustained pain freedom was achieved by 67% (10/15; CI95% 38%–88%).
Nausea, photophobia, and phonophobia disappeared at 2 hours in 54% (12/22; CI95% 32%–75%), 41.9% (13/31; CI95% 24% – 60%), and 40% (10/25; CI95% 21%–61%) participants, respectively. We also evaluated the disappearance of at least one associated symptom of nausea and/or vomiting, photophobia, and phonopho- bia (defined as disappearance of at least one symptom at 2 hours which was present at baseline. 66% (25/38; CI95% 49%–80%) of the participants experienced disappearance of at least one of the associated symptoms in the test treatment (1 participant who did not have any of the symptoms at baseline was excluded from the analysis). Furthermore, 69% (23/33; CI95% 51%–84%) participants experienced improvement in functional ability at 2 hours (only participants with functional disability at baseline were included in the analysis).
Two participants (5%) used medication within 2 hours of the test treatment, demonstrating compliance rate of 94%. Additionally, one participant (2%) started the test treatment over 60 minutes of attack onset, demonstrating compliance rate of 97% to treat early.
An additional highly important aspect of acute treatments of mi- graine from a clinical standpoint is efficacy across multiple attacks. Accordingly, to derive more stable estimates of long-term response to the treatment, a consistency analysis was conducted (excluding the training treatment). A total of 110 treatments (excluding the training treatment) were performed by the 39 participants (average of 2.8 treatments per participant) and included in the consistency analysis. This analysis demonstrated that 66% (26/39; CI95% 49% – 80%) of the participants experienced pain relief in at least 50% of their treated attacks, and 33% (13/39; CI95% 19%–50%) of the participants experienced pain-free in at least 50% of their treated attacks (Table 3 and Figure 2).
Improvement in migraine-related disability
Forty-two participants who completed the questionnaire both at baseline and at the end of treatment phase were included in the analysis. The change between the PedMIDAS scores at enrollment (37.1 [30.4]) and the end of the treatment phase (18.5 [26.8]) was 18.6 (23.4) with an interquartile range of 27.5.
DISCUSSION
This open-label study demonstrates that REN may offer a safe and tolerable acute treatment of migraine in adolescents. The findings of this study extend previous studies establishing the safety and efficacy of REN in adults with migraine. Specifically, the findings of this study show that the REN device is safe and well-tolerated. The incidence of device-related adverse events over multiple migraine attacks was low (2.2%), with no device-related serious adverse events. This rate compares favorably to the reported rates for current acute pharmacological treatments.
We also show a clinical benefit of REN for pain relief, pain-free, and disappearance of associated symptoms at 2 hours after acute migraine attack treatment. Pain relief and pain-free responses were sustained 24 hours after treatment. Furthermore, the data reveal consistent response rates from treatment to treatment, with no evidence of reduction in therapeutic benefits over time.
The 2-hour response rates to REN in adolescents (36% pain- free; 72% pain relief) were similar to those reported in adults. Importantly, over 66% of the patients achieved pain relief at 2 hours in more than half of their attacks and over 33% of the pa- tients achieved pain-free at 2 hours in more than half of their at- tacks, demonstrating consistent efficacy across multiple attacks. Our finding that pain relief and pain-free responses were sustained at 24 hours are also favorable, thought further studies are needed to assess the stability of this effect as these were exploratory out- comes which were assessed on a small number of subjects. We also measured response in at least one of the associated symptoms present at baseline for each attack. This analysis included all asso- ciated symptom and not only those considered most bothersome, yet it demonstrates that in 66% of the patients, REN treatments result in the disappearance of at least some of the associated symptoms. Furthermore, our findings demonstrated that REN has an effect on migraine related disability. Approximately 70% of patients reported improved function at 2 hours following REN treatment. Moreover, the average decrease in PedMIDAS scores observed in the current study is similar to the reduction shown for migraine preventive treatments in the pediatric population,19 suggesting that REN is also effective for improving patients’ quality of life.
This study was conducted both on adolescents with chronic migraine (at least 15 headache days per month, with migraine-like headaches on ≥8 days per month) and adolescents with non-chronic migraine. In this study, 11.7% (7/60) of the enrolled patients were adolescents with chronic migraine, of which 4/39 (10.3%) were included in the final analysis set. Although this small sample size precludes subgroup analyses, it suggests that REN may provide a drug-free treatment option for adolescents with migraine inde- pendently from the frequency of their migraines.
This study has several limitations. First, the efficacy results are not placebo controlled, which is specifically important in pediatric studies which typically show a higher rate of placebo response in adolescents than adults. However, even if accounting for the high placebo response rate of 55% observed for 2-hour pain relief in previous studies, the therapeutic gain in this study remains clinically meaningful. Second, this study was conducted on a small sample size. Further studies in a larger sample size are warranted.
CONCLUSIONS
This open-label study demonstrates that the incidence of device- related adverse events is very low, providing support that REN is well tolerated and safe. The study also shows that REN may be associated with clinically meaningful efficacy in adolescents with migraine. Therefore, REN may offer a novel alternative for current pharmacological treatments that combines efficient treat- ment with minimal side effects. The favorable safety profile and the clinical benefits introduce an alternative acute treatment that can be incorporated into usual care and may reduce medication use and holds the potential to improve the quality of life of adolescents with migraine.
