Remote Electrical Neuromodulation (REN) Relieves Acute Migraine: A Randomized, Double-Blind, Placebo-Controlled, Multicenter Trial
David Yarnitsky et al, Headache, 2019
Objective.—To assess the efficacy and safety of a remote electrical neuromodulation (REN) device for the acute treatment of migraine.
Background.—There is a significant unmet need for novel effective well-tolerated acute migraine treatments. REN is a novel acute migraine treatment that stimulates upper arm peripheral nerves to induce conditioned pain modulation – an endogenous analgesic mechanism in which conditioning stimulation inhibits pain in remote body regions. A recent pilot study showed that REN can significantly reduce headache. We have conducted a randomized, double-blind, sham-controlled study to further evaluate the efficacy and safety of REN for the acute treatment of migraine.
Methods.—This was a randomized, double-blind, sham-controlled, multicenter study conducted at 7 sites in the United States and 5 sites in Israel. Two hundred and fifty-two adults meeting the International Classification of Headache Disorders criteria for migraine with 2-8 migraine headaches per month were randomized in a 1:1 ratio to active or sham stimulation. A smartphone-controlled wireless device was applied for 30 45 minutes on the upper arm within 1 hour of attack onset; electrical stimulation was at a perceptible but non-painful intensity level. Migraine pain levels were recorded at baseline, 2, and 48 hours post-treatment. Most bothersome symptoms (MBS) were also recorded. The primary efficacy endpoint was the proportion of participants achieving pain relief at 2 hours post-treatment (improvement from severe or moderate pain to mild or none, or from mild pain to none). Relief of MBS and pain-free at 2 hours were key secondary endpoints.
Results.—Active stimulation was more effective than sham stimulation in achieving pain relief (66.7% [66/99] vs 38.8% [40/103]; therapeutic gain of 27.9% [CI95%, 15.6-40.2]; P < .0001), pain-free (37.4% vs 18.4%, P=.003), and MBS relief (46.3% vs 22.2%, P=.0008) at 2 hours post-treatment. The pain relief and pain-free superiority of the active treatment was sustained 48 hours post-treatment. The incidence of device-related adverse events was low and similar between treatment groups (4.8% [6/126] vs 2.4% [3/126], P=.499).
Conclusions.—REN provides superior clinically meaningful relief of migraine pain and MBS compared to placebo, offering a safe and effective non-pharmacological alternative for acute migraine treatment.
INTRODUCTION
Migraine is one of the most prevalent and disabling disorders, characterized by recurrent headache attacks with nausea, vomiting, photophobia, and phonophobia. Non-steroidal anti-inflammatory drugs (NSAIDs) and triptans, commonly used for acute migraine treatment, may be ineffective, poorly tolerated, contraindicated, and if used in excess, may lead to medication overuse headache and migraine chronification – profound barriers to optimal migraine care.Only 15.9% of the U.S. population with migraine use triptans, with an extremely high discontinuation prevalence of 55.2-81.5%. Thus, there is a great unmet need for alternative acute migraine treatments that are both effective and well tolerated.
Non-invasive neuromodulation is safe, well tolerated, and may have fewer adverse effects than drugs. Remote electrical neuromodulation (REN) is a novel acute migraine treatment that stimulates upper arm peripheral nerves to induce conditioned pain modulation (CPM) – an endogenous analgesia mechanism in which conditioning stimulation inhibits pain in remote body regions. The mechanism of REN and its potential use in migraine have been described in details in a recent pilot study. Presumably, REN activates descending inhibition pathways that originate in the periaque ductal gray (PAG) and in the rostral ventromedial medulla (RVM) which globally inhibit pain by the release of serotonin and noradrenalin (Fig. 1). The pilot study demonstrated that early treatment of migraine attacks with REN can significantly reduce headache. In this paper, we report the results of a randomized, double-blind, sham-controlled, multicenter pivotal study designed to evaluate the efficacy and safety of REN for the acute treatment of migraine.
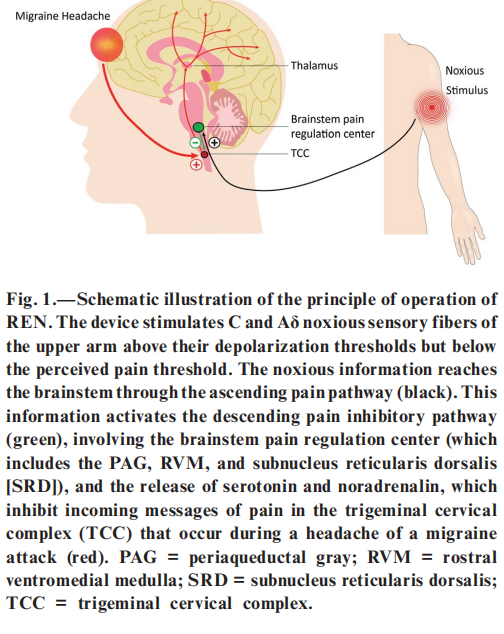
METHODS
Standard Protocol Approvals, Registrations, and Patient Consents. —We conducted the study at 7 sites in the United States and 5 sites in Israel. The study protocol was reviewed and approved by the appropriate institutional review board for each site and was conducted according to Good Clinical Practice and the Declaration of Helsinki guidelines. Before undergoing any study procedures, patients provided written informed consent. The first patient was enrolled in December 2017, and the last patient completed the double-blind phase of the study in October 2018. The study is registered with ClinicalTrials. The trial was conducted in accordance to the original protocol, which is available by request. Prior to unmasking, the statistical analysis plan was amended to explain in more details the analyses methods and procedures.
Study Design and Participants. —Patients with migraine with or without aura participated in this, prospective, randomized, double-blind, sham controlled, multicenter trial. Patients were eligible to participate if they were 18-75 years old, met the International Classification of Headache Disorders (ICHD 3-beta) criteria for migraine with 2-8 migraine headaches per month and <12 headache days per month, and were on either no or stable migraine preventive medications in the last 2 months prior to recruitment. Exclusion criteria were: (1) pregnancy, nursing, trying to conceive; (2) pure menstrual migraine (excluded because these attacks are often longer); (3) implanted electrical device(s); (4) treatment with OnabotulinumtoxinA in the prior month; (5) nerve blocks in the preceding 2 weeks; (6) current use of cannabis; (7) uncontrolled epilepsy; (8) receiving parenteral infusions for migraine in the preceding 2 weeks; (9) other significant pain, medical or psychiatric illness that in the opinion of the investigator may confound the study assessments; (10) unable to use a smartphone; and, (11) previous experience with REN in clinical trials for migraine. The rationale for the exclusion of botox, nerve blocks, and IV infusion was to ensure there would be enough attacks during the run-in phase of the study. Due to the temporal pattern of botox treatment, in which the positive effect of the treatment shows a tendency to wane between the first and third months after the treatment, we specifically excluded those who used botox 1 month prior to enrollment.
Stimulation Device. —The REN device (Nerivio Migra®, Theranica Bio-Electronics Ltd., Israel) is a wireless wearable battery-operated stimulation unit controlled by a smartphone software application. The device is applied for 45 minutes on the lateral upper arm between the bellies of the lateral deltoid triceps, so that it will mainly stimulate small skin nerves. The rationale for stimulating the arm and the underlying mechanism of action are described in details elsewere. The active device produces a proprietary electrical signal comprising a modulated symmetrical biphasic square pulse with a modulated frequency of 100-120 Hz, pulse width of 400 μs, and output current up to 40 mA (adjusted by the participant). Although the pulse stimulates C and Aδ noxious sensory fibers above their depolarization thresholds, the stimulation energy is low enough to maintain the overall sensory experience below perceptual pain threshold. The sham device differed from the active device in the pulse frequency which was~0.083 Hz and the pulse width which was 40-550 μs (modulated), aimed to induce a solid and perceptible sensation similar to the active device, but with a sufficient low frequency to prevent any modulation of nociceptive processes. The parameters of the sham and active devices were chosen following a pilot study which used several stimulation programs in a crossover design.The stimulation intensity of both the active and sham devices was adjusted by the users using the app.
Randomization, Masking, and Blinding. —Before each site initiation, randomization schemes of blocks of 8 participants per site (4 active and 4 sham) were developed. Group allocation was concealed by randomization lists for each smartphone operating system (Android or iOS) prepared by the statistician of the data monitoring committee (DMC), which included the device MAC address (the unique device identification number) and an assigned randomization number identifier. Participants were randomized based on the type operating system of their phones (Android or iOS) and their arbitrary order of arrival to the randomization visit. To assess blinding, participants were asked at the end of study of their presumed group assignment (active, sham, do not know). The participants, investigators, and clinic personnel were unaware of group assignment until the completion of the double-blind treatment phase, when the database was opened. Each device was programmed with 1 of 2 versions of firmware that deliver either active or sham electrical stimulation (see above for detailed electrical properties). In all other aspects, the active and the sham conditions were kept identical.
Procedures. —After enrollment, participants were trained to use the electronic diary application, installed on their own smartphones, and then completed a 2-4 weeks migraine diary (“run-in” phase). Eligible participants were then randomized in a 1:1 ratio to either active (active group) or sham stimulation (sham group), in a double-blind manner. During the randomization visit, participants were trained to use the device assigned to them (either active or sham), including finding the optimal individual stimulation intensity level (perceptible but not painful). Participants treated their migraine attacks at home for 4-6 weeks (“double-blind treatment” phase), with their optimal stimulation intensity, as soon as possible after migraine headache began and always within 1 hour of symptom onset. Participants were instructed to avoid taking rescue medications within 2 hours post-treatment. Pain scores (none, mild, moderate, or severe) were recorded at baseline, 2, and 48 hours post-treatment. Migraine-associated symptoms including nausea, photophobia, and phonophobia were recorded at the time of treatment. Participants declared their most bothersome migraine symptom (MBS) for each treated attack and reported absence and presence of all associated symptoms (including their MBS) at baseline and 2 hours post-treatment. At 2 hours, participants also subjectively reported whether they feel that a significant MBS relief was achieved. At 2 and 48 hours, participants reported if, when and what type of acute medication was used.
Outcomes. —The primary efficacy endpoint was the proportion of participants who achieved pain relief at 2 hours post-treatment in the test treatment, defined as improvement from severe or moderate pain to mild or none, or improvement from mild pain to none. The secondary efficacy endpoints pain free (improvement from mild, moderate, or severe pain to none), MBS relief, pain and MBS relief, and MBS freedom at 2 hours post-treatment. Exploratory endpoints included 48-hour sustained pain-free and pain relief responses (see Glossary). An additional exploratory endpoint, conducted on multiple attacks, was within-subject consistency, defined as the proportion of participants achieving pain relief at 2 hours post-treatment in at least 50% of their treated attacks. Pain relief at 2 hours post-treatment as function of baseline pain level was also explored.
Statistical Analysis. —A sample size of 234 participants (117 per treatment arm) was determined to provide 80% power to demonstrate statistical significance of .05 for the primary endpoint, assuming a sham pain relief rate of 32% and a therapeutic gain of 18%. Accounting for a ~13% discontinuation rate, 270 participants were initially planned to be enrolled. A planned blinded interim analysis, performed and reviewed by an independent external DMC, confirmed the sample size calculations. However, an unanticipated ~16% rate of incomplete data was revealed, and thus the required total number of participants was increased to approximately 300.
The intention-to-treat (ITT) population included all the participants who underwent randomization and was used for safety analyses. To avoid treating recurrent headaches, the efficacy analyses were performed on treated attacks preceded by at least 48 headache-free hours. The first reported treatment of each participant was considered a training treatment and was only included in the safety analyses. The efficacy evaluation was based on the first fully treated (30-45 minutes) attack with 2 hours post-treatment assessment following the training treatment (test treatment). Statistical efficacy analyses (active vs sham) were conducted in the modified intention-to-treat (mITT) population, defined as all randomized participants who had performed a test treatment within 1 hour from symptom onset. The analyses were performed on all participants in the mITT, regardless of their efficacy results in the training treatment. Use of rescue medication before the 2 hours assessment was considered a treatment failure. Migraine attacks presented upon awakening were excluded from the analyses since the time of onset was unknown. As a sensitivity analysis, the primary endpoint was also evaluated with missing data imputed using a worst-case scenario approach, in which all treatments with missing pain level data in the active group were considered failures and all treatments in the sham group with missing pain level data were considered successes. MBS relief was assessed using a subjective yes or no question: “was a significant MBS relief achieved?”. Participants who did not provide data on associated symptoms were excluded from MBS analyses. Multiple attacks were used for one of the exploratory outcomes assessing within-subject consistency.
For continuous variables, mean, and standard deviation are provided. For categorical variables, the number and percentage of patients in each category are provided. The efficacy and safety outcomes were evaluated using two-tailed chi-squared test, two-tailed Fisher’s exact test or the Breslow-Day test, as appropriate. Specifically, the primary and secondary efficacy endpoints were evaluated using two-tailed chi-squared test. The continuous variable of age was compared using an independent-sample two-tailed t-test. The assumptions of normality and of homogeneity of variance of the age variable were verified using the normal quantile–quantile (Q–Q) plot and Levene’s test of equality of variances (P = .605), respectively. A value of P < .05 was used to identify statistically significant effects. To control for type I error, a hierarchical testing procedure was applied in which the secondary endpoints were tested only if the primary endpoint was significant. The Benjamini–Hochberg step-up method was used to correct for multiple comparisons in the analyses of the secondary efficacy endpoints. All data were analyzed using SAS® v9.4 (SAS Institute Inc., Cary, NC). All authors had full access to all study data.
RESULTS
Participants. —This study was conducted from December 17, 2017 to October 7, 2018. Two hundred and ninety-six participants were enrolled; 252 were randomized after the “run-in” phase, 126 were assigned to active group, and 126 to sham group (see Fig. 2 and Table 1 for patient characteristics). Of the 44 nonrandomized, 33 failed to meet the run-in criteria, 9 withdrew from the study, and 2 were withdrawn by the investigator due to migraine misdiagnosis. Of the 33 participants who failed to meet the runin criteria, 21 participants were ineligible due to insufficient number of attacks (less than two attacks), 11 participants were ineligible due to noncompliance with migraine attack diary reporting requirements, and 1 participant was ineligible due to change in preventive medication. Among the 252 randomized participants, 7 withdrew from the study, 3 (1 sham, 2 active) due to intolerance to the stimulation, and 4 (2 sham, 2 active) were lost to follow up. Two hundred and thirty-seven participants completed at least one treatment (the training treatment) and 203 participants completed the test treatment. Two hundred and two participants (99 in the active group and 103 in the sham group) started the test treatment within 1 hour from symptom onset and reported pain level at 2 hours, forming the mITT population with evaluable data. Thirty-seven participants who did not complete the test treatment, due to insufficient migraine attacks (n = 18), participant’s decision not to use the device (n = 4), other (n = 6), or unknown (n = 9), were excluded from the efficacy analyses.
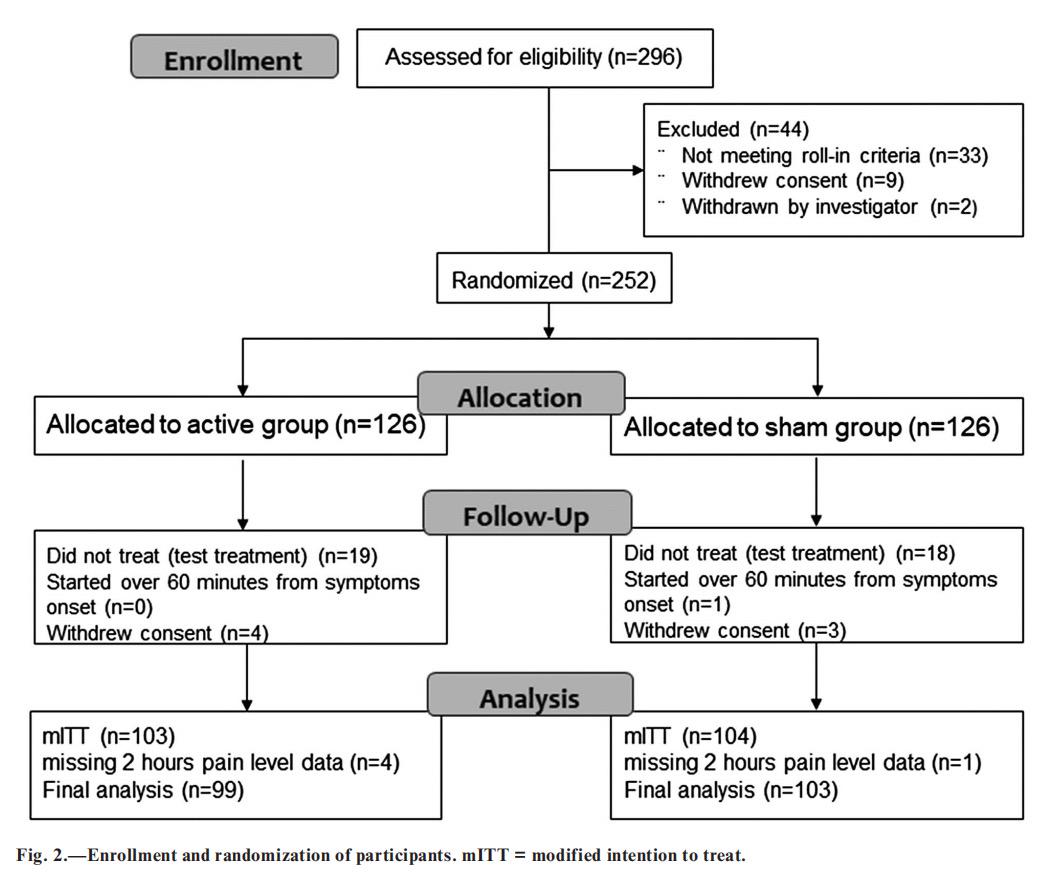
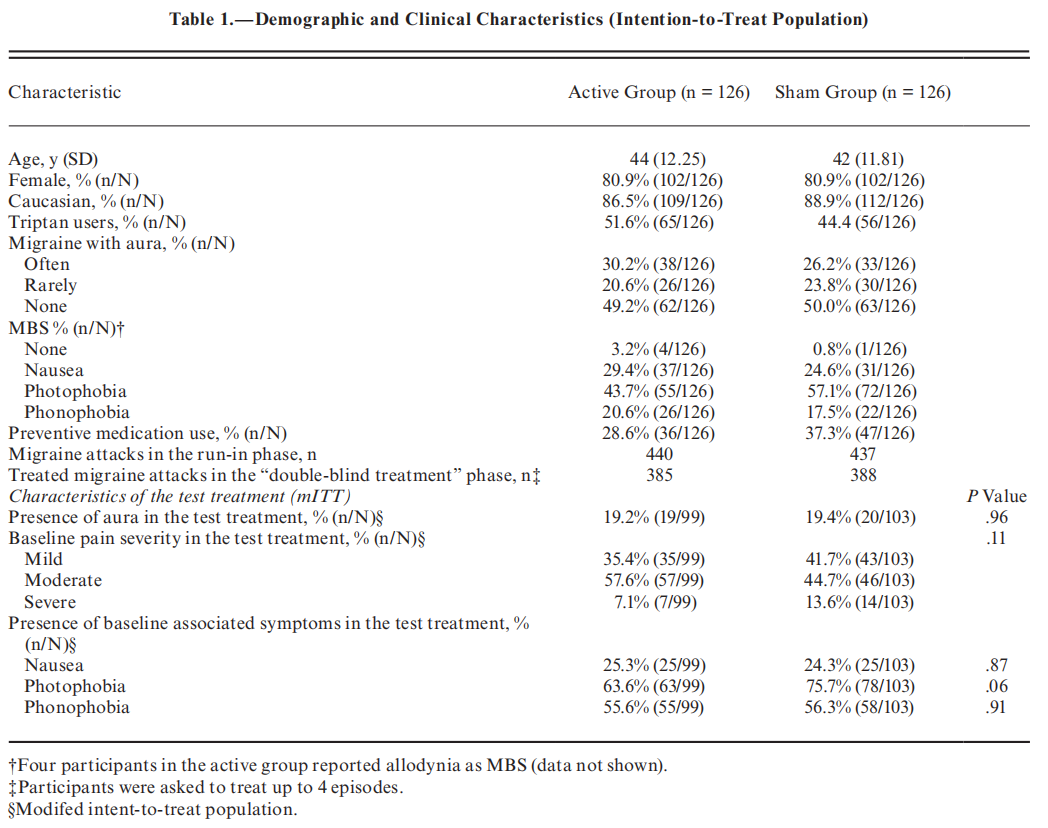
The demographic and clinical characteristics were similar between treatment groups (Table 1). The characteristics of treated migraine headaches were comparable to those reported in previous migraine studies.
Efficacy Outcomes. —In the active group, 66.7% participants (66/99; CI95% 56.48-75.82) achieved pain relief at 2 hours post-treatment for the test treatment, compared to 38.8% (40/103; CI95%, 29.39-48.94) in the sham group, a therapeutic gain of 27.9% (CI95%, 15.6-40.2; P < .0001; Fig. 3A and Table 2). Similar results were found when missing data were imputed using a worst-case scenario (66/103=64.1% for active vs 41/104=39.4% for sham, P < .0005). Importantly, pain relief at 2 hours post-treatment did not depend on the site (Breslow-Day test; P=.9706). Furthermore, more participants in the active group were pain-free at 2 hours post-treatment (37.4% [37/99; CI95% 27.85-47.67]) compared with the sham group (18.4% [19/103; CI95% 11.49-27.30]; a therapeutic gain of 19.0%; P=.003, adjusted; Fig. 3A and Table 2). Rescue medication within 2 hours post-treatment was used by 1% of the participants in the active group and 3.8% in the sham group (P=.190).
The active treatment was also significantly more effective than sham for MBS relief (46.3% [44/95] vs 22.2% [22/99]; CI95% 36.02-56.85 and 14.48-31.69 respectively; P = .0008, adjusted) and for combined pain relief & MBS relief (40.0% vs 15.2%; CI95% 30.08-50.56 active and 8.74-23.76 sham; P = .0004, adjusted) at 2 hours post-treatment (Fig. 3B and Table 2). There was no statistically significant difference between treatment groups in the 2 hours post-treatment MBS free response (40.7% [33/81] vs 36.4% [32/88]; CI 29.95-52.23 active and 26.37-47.31 sham; P = .559, adjusted; Fig. 3B and Table 2).
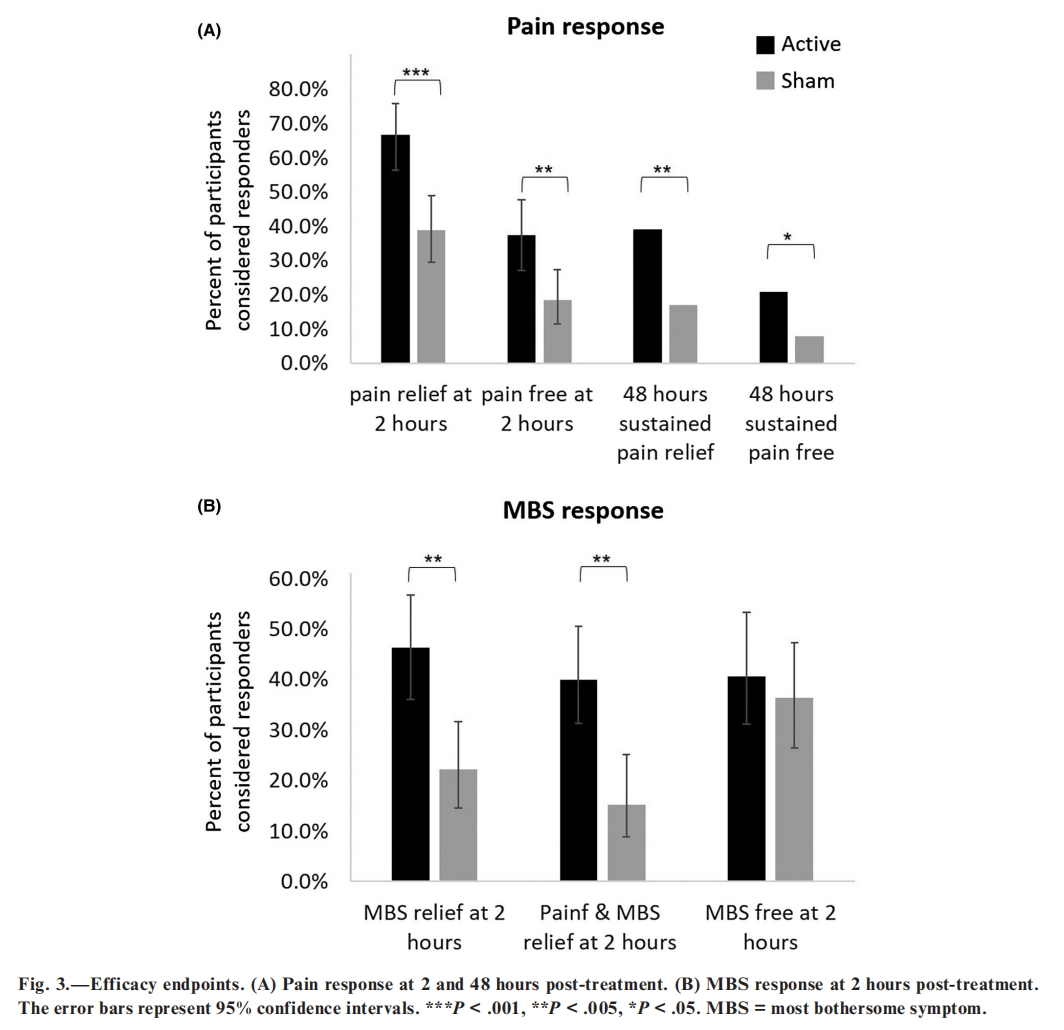
The 2-hour pain relief and pain-free superiority of the active treatment were sustained 48 hours post treatment. Sustained pain relief at 48 hours post treatment was achieved in 34 of 87 (39.1%) participants in the active group, and in 15 of 89 (16.9%) participants in the sham group (P = .001; Fig. 3A). Sustained pain free at 48 hours was achieved in the active group by 18 of 87 (20.7%) participants, and in the sham group by 7 of 89 (7.9%) participants (P = .015; Fig. 3A).
During the double-blind treatment phase, participants in the active group treated an average of 3.5 attacks per participant vs an average of 3.6 attacks per participant in the sham group. All treated attacks (excluding the training treatment) were used for within-subject consistency assessment. Pain relief at 2 hours post-treatment for at least 50% of treated attacks was higher in the active group compared to the sham group (62.6% [62/99] vs 45.6% [47/103], P = .015; Table 2).
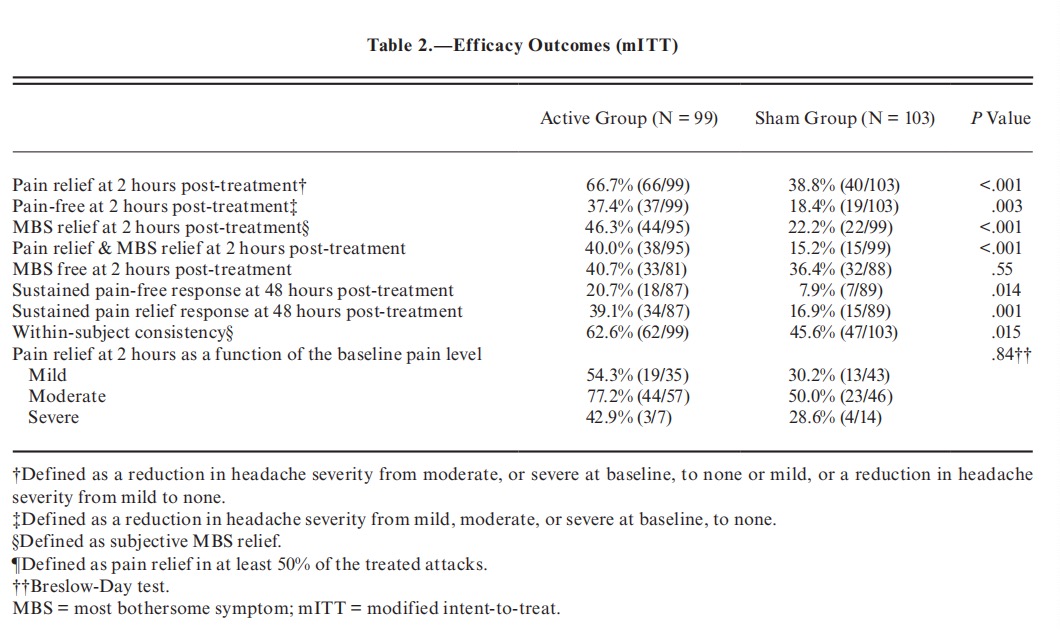
The primary end-point was also evaluated as a function of baseline pain intensity. The interaction between baseline pain intensity and response rate was not found significant (P = .841; Table 2), indicating that the treatment effect was similar across baseline pain intensity levels.
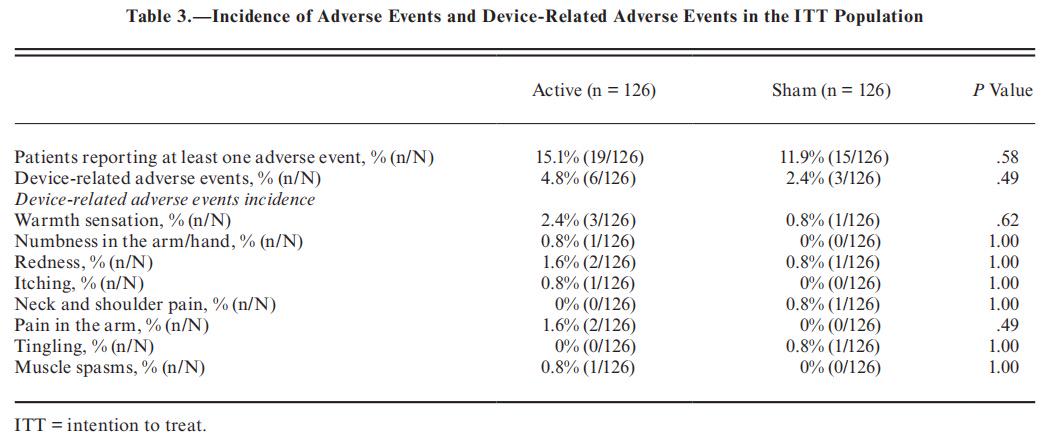
Safety. —Safety analyses were performed on all 252 participants (ITT population). The percentage of participants experiencing at least one adverse event was 13.5% (34/252) and was comparable across treatment groups (15.1% [19/126] active and 11.9% [15/126] sham, P = .581). device-related adverse events were reported during 773 treatments (2.7%), in the active group, and 9 in the sham group. The incidence of device-related adverse events was low (3.6%) and similar between treatment groups (6/126 [4.8%] vs 3/126 [2.4%]; P = .499). Device-related adverse events included warmth sensation, temporary arm/hand numbness, redness, itching, tingling, muscle spasm, and pain in the arm, shoulders, or neck (Table 3). All device-related adverse events were mild, resolved within 24 hours and did not require medical treatment. There were no device-related serious adverse events, no unanticipated adverse device effects and none of the participants withdrew from the study due to adverse events.
Blinding. —Upon treatment completion, participants were asked of their presumed group assignment (active, sham, do not know). 44.6% (50/112) in the active group and 41.9% (49/117) in the sham group did not know. In the active group, 23.2% (26/112) believed they had received the active device, and 32.1% (36/112) the sham. In the sham group, 50.4% (59/117) believed they had received the sham device and 7.7% (9/117) the active. Two types of analyses were performed to examine whether the treatment effect depends on the perceived assignment. The Cochran–Mantel–Haenszel test (CMH) was used for the analysis of the data stratified to perceived assignment (P = .004, mITT analysis set). The low P value suggests that even when controlling for the participants responses (perceived assignment), the treatment effect (active vs sham) remained significant, indicating that the perceived assignment does not explain the difference between the active and sham groups. Thus, the association between treatment and response remained strong after adjusting for perceived assignment. The Breslow-Day test was used to assess whether the difference between active and sham groups depends on the perceived assignment (P = .911). The high P value of this analysis indicates that the interaction is not significant and thus the difference between the active and sham groups is not affected by the perceived assignment.
DISCUSSION
We found a statistically significant, clinically important benefit of REN compared to sham treatment for pain relief, pain-free, MBS relief, and the combination of MBS and pain relief at 2 hours after acute migraine attack treatment. Pain relief and painfree responses were sustained 48 hours after treatment in more participants in the active than in the sham group. The benefits were maintained in subsequent attacks and were independent of pain levels prior to treatment. This trial is one of the largest ranomized, double-blind, sham-controlled device studies for acute migraine.10 It confirms the favorable effcacy and safety outcomes that have been previously reported using this device.
REN likely utilizes a descending endogenous analgesia mechanism to control migraine pains (for further details see Yarnitsky et al). While the descending pain modulatory system is well characteized, this natural mechanism has not been utilized for non-pharmacological pain treatments, except for invasive spinal cord stimulation that is believed to activate this system. Engagement of this descending network is believed to induce CPM (a human paralel of the animal lab-based diffuse noxious inhibitory control effect) to inhibit headache pain by peripheral nociceptive information which is below the perceived pain threshold.
To our knowledge, this device presents the first attempt to exploit CPM for non-pharmacological, non-invasive treatment of pain. This method is distinct from current neuromodulation treatments for migraine. When the head or neck is stimulated it triggers a local stimulus–response that relies on the gate control theory. Transcutaneous magnetic simulation (TMS) is believed to inhibit cortical spreading depression, and vagal stimulation is believed to active autonomic–somatic inhibitory interaction. We utilized upper arm stimulation to produce pain inhibition. This is a discreet, practical, and convenient treatment that enables patients to continue with daily activities.
The efficacy of REN is superior to that reported for other neuromodulation devices intended for acute migraine treatment, with a 2 hours pain relief therapeutic gain of 27.9% compared to 5.0% of single transcranial magnetic stimulation and 13.2% of non-invasive vagus nerve stimulation (nVNS). In fact, the study on nVNS for acute treatment of migraine did not meet its primary endpoint of pain free at 2 hours post-treatment (therapeutic gain of 10.7%), which was successfully achieved in the current study, with a therapeutic gain of 19.0%. The therapeutic gain of pain free at 2 hours in the current study was also higher than the therapeutic gain of 10% reported for external trigeminal nerve stimulation. To compare the efficacy of REN to that of triptans, we analyzed the pain relief and pain-free responses at 2 hours post-treatment in attacks with moderate or severe baseline pain intensity. At 2 hours post-treatment, 73.4% of the participants in the active group and 45.0% in the sham group achieved pain relief (P = .001); and 28.1% and 10.0%, respectively, achieved pain freedom (P = .011). These findings suggest that REN is equivalent to triptans, which have a 42-76% pain relief response rate, and ~18-50% pain-free response at 2 hours. These findings also point to non-inferiority of REN compared to newly developed gepants, which have a 2 hours pain freedom therapeutic gain of 16.6-17.6%. Limitations of these comparisons are that they are not a head to head trials with REN, the design, and number of subjects vary between studies, and that participants treated early within 1 hour of symptom onset.
REN is effective for pain relief and freedom as well as for MBS relief, but not for MBS-free response at 2 hours. Possibly, the reduced effect on the non-nociceptive features results from the engagement of parallel mechanisms unrelated to the pain circuit. Alternatively, the lack of significance could be attributed to a high placebo effect in the sham group, which is consistent with previous studies assessing MBS-free response.
Adverse event incidence was low; mainly reports of sensation of warmth, redness, and numbness of the arm/hand. The safety profile of REN is favorable compared to triptans and to new pharmacological agents, such as centrally acting serotonin (5-HT1F) agonists that lacks vasoconstrictive activity. No cephalic electrical neuromodulation may have fewer side effects than other available cephalic neuromodulation treatments.
The current study has several limitations. First, there was a low rate of severe baseline pain intensity and high rate of mild pain intensity, presumably due to the early treatment. Yet, the rates of pain relief were as high for attacks treated at a moderate pain level, as for those treated at a mild pain level. Second, we did not study the efficacy of the device at intervention periods over 1 hour of symptoms onset. Finally, selecting an appropriate sham device for successful blinding in neuromodulation studies in migraine is a great challenge. However, in the current study the sham device produced a solid perceivable stimulus. As in other neuromodulation studies in migraine, the placebo effect was higher than drug trials. Yet, the therapeutic gain in the current study was impressive and was not significantly affected by the participants’ treatment-assigned response, providing acceptable evidence that REN treatment is effective and safe as an acute treatment for migraine.
CONCLUSIONS
Our findings suggest that REN is an effective acute migraine treatment with a favorable safety and tolerability profile. REN may be an alternative acute migraine treatment with comparable or superior efficacy to commercially available neuromodulation devices. REN has the potential to increase patient adherence, improve migraine management, and improve the health and quality of life of people with migraine.
